Salmonella Typhimurium infection blocks ER-phagy
To investigate the possible impact of Salmonella infection on ER-phagy, we generated a HEK293A cell line stably expressing a previously published doxycycline-inducible ER-phagy reporter containing an ER signal sequence, followed by the fluorescent proteins RFP and GFP, and the ER retention sequence KDEL (ss-RFP-GFP-KDEL)7. Upon doxycycline treatment and ER-phagy induction, the fluorescent ER-associated reporter is sequestered inside autophagosomes and cleaved upon lysosome fusion. Unlike GFP, RFP is relatively resistant to lysosomal pH and hydrolases22. As such, a ~25-kDa fragment corresponding to RFP can be detected by western blot upon activation of ER-phagy. Similarly, the same reporter can be used to measure ER-phagy by fluorescent microscopy due to the quenching of GFP fluorescence at lysosomal acidic pH (Fig. 1A). As a result, ER-phagy also can be monitored as an increase in RFP positive structures (lysosome-associated ER-phagy probe) compared to the dual positive probe signal. We tested the reporter by activating ER-phagy through amino acid starvation7 or Torin-1 (a potent autophagy inducer) treatment. As a negative control, we confirmed that ss-RFP-GFP-KDEL did not get cleaved in autophagy-deficient FIP200 KO cells (Supp Fig. 1A).
Amino acid starvation for 6 h is sufficient to induce ER-phagy. To determine if Salmonella infection impacted ER-phagy, we added Salmonella to the amino acid starvation media for the final 1 or 3 h of starvation. Infection with Salmonella for either 1 or 3 h significantly reduced the production of the free RFP fragment when compared to 6 h of amino acid starvation in the absence of Salmonella (Fig. 1 B C), indicating inhibition of ER-phagy upon infection. RFP processing was inhibited by Salmonella when normalized to either actin (Fig. 1C) or total ss-RFP-GFP-KDEL reporter (Supp Fig. 1B). We expanded our analysis of ER-phagy regulation by Salmonella in the HCT116 cell background, which were engineered to stabily express ss-RFP-GFP-KDEL and observed a similar repression of ER-phagy (Supp Fig. 1C and Supp Fig. 1D). Moreover, both HEK293A and HCT116 cells expressing a HaloTag-based pulse-chase reporter for ER-phagy measurement23, showed decreased ER-phagy levels upon Salmonella infection, further corroborating a role for Salmonella in repressing ER-phagy (Supp Fig. 1E and Supp Fig. 1F). Interestingly, Salmonella infection failed to block non-selective autophagy as shown by the degradation of p62/SQSTM1, a cargo receptor regularly used as an index for general autophagic degradation and LC3B-II production, the faster electrophoretic form of LC3B that is produce during autophagy initiation by LC3B lipidation24,25 (Supp Fig. 1G and Supp Fig. 1H). Similarly, the general autophagy reporter mCherry-eGFP-LC3B24, showed no significant differences (RFP puncta formation) between infected and non-infected cells during starvation (Supp Fig. 1I and Supp Fig. 1J). Furthermore, the commercial autophagy detection kit, CYTO-ID, confirmed that non-selective autophagy was not inhibited by Salmonella infection during starvation, suggesting Salmonella specifically targets ER-phagy (Supp Fig. 1K). Importantly, Bafilomycin A1 (BafA1) treatment, a potent inhibitor of autophagosome maturation and cargo degradation, completely blocked free RFP production when cells were starved in the presence of Salmonella, indicating that Salmonella did not increase the turnover rate of autolysosomes (Supp Fig. 1L). The ss-RFP-GFP-KDEL probe can be used to visualize ER-phagy by fluorescence microscopy, where ER-phagy induction is observable through detection of RFP puncta formation. Infection with Salmonella resulted in a significant decrease in ER-phagy induction compared to cells that were not infected (Fig. 1D, E), which was consistent with our western blot analysis. Similar results were observed when we repeated the experiment using the ss-RFP-GFP-KDEL probe in HCT116 cells (Supp Fig. 1M, N), and the HaloTag-based ER-phagy reporter in HEK293A and HCT116 (Supp Fig. 1O, P). We next measured the protein levels of endogenous ER-phagy cargo receptors known to be involved in starvation-induced ER-phagy, namely TEX264 and FAM134B. Because these receptors link the ER to the autophagosome membrane they are ultimately degraded making their protein levels inversely correlated with ER-phagy levels6,7. We measured the endogenous level of each receptor during ER-phagy inducing conditions in the absence and presence of Salmonella and found both TEX264 and FAM134B levels to be significantly higher upon infection, indicating that their degradation is blocked upon Salmonella infection (Fig. 1F, G). Conversely, non-selective p62/SQSTM1 autophagic degradation was similar between infected and non-infected samples. Similar results were observed when these experiments were repeated in HeLa cells (Supp Fig. 1Q). Additionally, Salmonella infection was also able to prevent the degradation of the ER-phagy receptors: FAM134A, FAM134C and RTN3L, further demonstrating the ability of Salmonella to block ER-phagy (Supp Fig. 1R, S). Collectively, these findings indicate that Salmonella infection specifically inhibits ER-phagy, but not non-selective autophagy.
FAM134B is targeted by Salmonella to block ER-phagy
The ER-phagy receptor proteins FAM134B and TEX264 have been previously reported to be targeted by invasive pathogens to disrupt ER morphology and promote infection19. To investigate this possibility, we generated CRISPR KO cell lines of FAM134B and TEX264 (Supp Fig. 2A). As expected, deletion of FAM134B and TEX264 severely decreased ER-phagy (Fig. 2A), which is in line with prior reports7. Consistent with our previous findings, WT cells infected with Salmonella showed a significant decrease in the levels of free RFP compared to uninfected cells during ER-phagy inducing conditions. However, Salmonella infection failed to show a significant difference in ER-phagy levels between infected and uninfected FAM134B KO cells. In contrast, Salmonella was still capable of repressing ER-phagy in TEX264 KO cells (Fig. 2A, B). Immunofluorescence microscopy using the same experimental setup showed a dramatic decrease in RFP puncta formation when ER-phagy was induced in Salmonella-infected WT and TEX264 KO cells. However, RFP puncta formation in FAM134B KO cells infected with Salmonella during ER-phagy induction failed to decrease to the same extent, displaying significant differences (Fig. 2C, D). To further analyze the requirement of FAM134B for Salmonella-mediated ER-phagy repression we created stable cell lines expressing an ER-phagy HALO reporter in WT and FAM134B KO cells. Using this reporter, we confirmed the inability of Salmonella to inhibit ER-phagy in the FAM134B KO background (Supp. Fig. 2B), consistent with the proteolytic cleavage of our ss-RFP-GFP-KDEL reporter (Fig. 2A). These results suggest that Salmonella infection primarily targets FAM134B to inhibit ER-phagy.

A WT, FAM134B KO, TEX264 KO and FIP200 KO HEK293A cells stably expressing the ss-RFP-GFP-KDEL were either kept in nutrient rich media, starved for AA for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Actin was used as a loading control. AA, amino acids; ST, Salmonella Typhimurium. B Normalized Free RFP-Actin ratio from cells in (A). Data are presented as mean values ± SD of seven biological experiments. ANOVA, *P < 0.05; ***P < 0.001; ns, no significance. C Cells from (A) were either AA starved for 6 h, or AA starved for 3 h, followed by ST infection for 3 h in AA starvation media, fixed and imaged by confocal microscopy. NR, Nutrient Rich. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red. Scale bar 5 μm. D Average number of RFP puncta per cell from (C) were quantified. Data are presented as mean values ± SD of 5 biological experiments. The averages were calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. E WT, FAM134B KO, TEX264 KO and FAM134B KO cells transfected with WT FAM134B were infected with ST. Bacterial content was determined through a colony-forming unit (CFU). Data are presented as mean values ± SD of three biological experiments. ANOVA, **P < 0.01; ***P < 0.001. F FAM134B KO cells transfected with either FAM134B WT or FAM134B LIR mutant were kept in nutrient rich media, starved for AA for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Actin was used as a loading control. G Cells from (F) were fixed and imaged by confocal microscopy. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red; Fam134B, far red. Scale bar 5 μm. H Normalized Free RFP-Actin ratio from cells in (F) and average number of RFP puncta per cell from (G) were quantified. Data are presented as mean values ± SD of five biological experiments. The average number of RFP puncta was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001; ns, no significance. Source data are provided as a Source Data file.
Next, we sought to determine if Salmonella-mediated ER-phagy inhibition impacted intracellular bacterial viability. To this end, we performed a colony-forming unit (CFU) assay in WT, FAM134B KO, TEX264 KO and FAM134B KO cells transfected with FAM134B WT. Analysis of Salmonella viability at 4 h post-infection revealed that FAM134B KO cells had significantly higher number of viable internalized bacteria, suggesting either a defect in the clearance of Salmonella or increased growth when compared to WT and TEX264 KO cells. Furthermore, FAM134B KO cells reconstituted with FAM134B recovered similar levels of Salmonella viability as parental cells (Fig. 2E), indicating FAM134B, but not TEX264, is important for Salmonella growth and survival.
ER-phagy receptors couple the ER to the autophagosomal membrane through LIRs (Fig. 1A). We next sought to determine if the impact of FAM134B on Salmonella growth was due to ER-phagy induction or another uncharacterized autophagy-independent function. To this end, we transfected FAM134B KO cells with either FAM134B WT or FAM134B LIR mutant and performed the ER-phagy reporter RFP processing assay. Intriguingly, we observed no significant difference between infected and uninfected ER-phagy induced cells when the FAM134B LIR mutant was expressed. Conversely, cells expressing FAM134B WT showed a significant decreased in free RFP production when infected with Salmonella (Fig. 2F, H). Consistent with our western blot analysis, we also observed that the decrease in the number of RFP puncta in the FAM134B LIR mutant cells triggered by Salmonella infection was not as pronounced as the one in infected cells expressing FAM134B WT (Fig. 2G, H). When we quantified Salmonella viability by CFU in HCT116 cells, results showed that the expression of the FAM134B LIR mutation resulted in a significant increase in the number of Salmonella compared to cells expressing FAM134B WT (Supp Fig. 2C). Together, these findings suggest that FAM134B plays a crucial role in limiting Salmonella burden and that this role is connected to FAM134B ability to induce ER-phagy.
FAM134B oligomerization is hindered by Salmonella infection
FAM134B promotes ER membrane scission and ER-phagy through its ability to oligomerize9. Therefore, we hypothesized that Salmonella infection might prevent ER-phagy by repressing FAM134B oligomerization. To test this hypothesis, FAM134B-FLAG was immunoprecipitated (IP) after 3 h of ER-phagy induction in the presence and absence of Salmonella infection. Consistent with previous reports, FAM134B oligomers were relatively resistant to denatured conditions and are observed by western blot in a distinct, slow migrating band9,14. Notably, Salmonella infection significantly reduced the formation of FAM134B oligomers (Fig. 3A). Similar results were observed when we repeated the experiment in HCT116 cells (Supp Fig. 3A). Furthermore, amino acid starvation-induced FAM134B puncta formation was dramatically reduced by Salmonella infection, revealing Salmonella blocks ER membrane scission, which is driven by FAM134B oligomerization (Fig. 3B). These results indicate Salmonella may block ER-phagy by preventing the oligomerization of FAM134B. If Salmonella represses FAM134B by preventing oligomerization, then forcing FAM134B oligomerization would be predicted to bypass Salmonella-mediated ER-phagy reduction. To this end, we generated a FAM134B G216R mutant, a naturally occurring mutation that resides in FAM134B reticulon-homology domain and has been described to dramatically enhance FAM134B oligomerization9,26. We transfected either FAM134B WT or FAM134B G216R in FAM134B KO cells and measured ER-phagy through the RFP processing assay. As expected, cells transfected with FAM134B WT exhibited reduced ER-phagy under stimulated conditions when infected with Salmonella (Fig. 3C, D). However, cells transfected with FAM134B G216R showed no significant difference in ER-phagy rates in the presence or absence of Salmonella, indicating that Salmonella inhibits ER-phagy upstream, or at the level, of FAM134B activation. Analysis of ER-phagy by immunofluorescence under the same conditions, showed that FAM134B G216R largely prevented ER-phagy repression by Salmonella, compared to FAM134B WT (Fig. 3E, F). Additionally, Salmonella infection failed to decrease FAM134B G216R oligomerization (Supp Fig. 3B), showing a dramatic increase in self-interaction and oligomerization as previously reported9. Together, these experiments support a model in which Salmonella regulates ER-phagy through repression of FAM134B activity.

A FAM134B KO HEK293A cells transfected with FAM134B-FLAG were starved for AA for 3 h in the absence or presence of ST. FLAG was immunoprecipitated. Data are presented as mean values ± SD of five biological experiments. Student’s t test, ***P < 0.001. AA amino acids; ST, Salmonella Typhimurium. B WT cells were incubated with BafA1 and starved for AA for 3 h in the presence or absence of ST. Fam134B puncta formation was observed by confocal microscopy. NR, Nutrient Rich; BafA1, Bafilomycin A1. Data are presented as mean values ± SD of five biological experiments. The average number of Fam134B puncta was calculated from a minimum of 100 cells. Representative images are shown. Fam134B, red; DAPI, blue; ST, green. Scale bar 5 μm. ANOVA, *** P < 0.001. C FAM134B KO cells were transfected with either FAM134B WT or FAM134B G216R and starved for AA for 6 h or starved for 3 h, followed by ST infection for 3 h in AA starvation media. ER-phagy was measured by ss-RFP-GFP-KDEL processing. Actin was used as a loading control. D Normalized Free RFP-Actin ratio from cells in (C). Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001; ns, no significance. E FAM134B KO were transfected with either FAM134B WT or G216R and starved for AA for 6 h or starved for 3 h followed by ST infection for 3 h in AA starvation media. Representative images are shown. ss-RFP-GFP-KDEL, yellow; Free RFP, red; Fam134B, far red. Scale bar 5 μm. F Average number of RFP puncta per cell from (E) were quantified. Data are presented as mean values ± SD of five biological experiments. The average number of RFP punctuation was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. Source data are provided as a Source Data file.
The Salmonella effector SopF blocks ER-phagy
In order to promote invasion and replication, Salmonella expresses two type-III secretion systems, which deliver multiple bacterial effectors into the host cells27. We hypothesized that one of these effectors might be involved in Salmonella-mediated ER-phagy inhibition. To test our hypothesis, we repeated the ss-RFP-GFP-KDEL processing assay infecting them with different Salmonella effector mutants. Among the mutants tested, Salmonella defective for the phosphoinositide-binding effector SopF showed the most complete and consistent loss of ER-phagy regulation (Supp Fig. 4A). Analysis of ER-phagy by western blot and immunofluorescence in our reporter cells showed that SopF-deficient Salmonella was unable to supress ER-phagy compared to WT Salmonella infected controls under stimulated conditions (Fig. 4A–D). Together, these experiments indicate that SopF is necessary for Salmonella-induced ER-phagy repression. Next, we sought to determine if the expression of the SopF effector was sufficient to inhibit ER-phagy. To this end, ER-phagy reporter cells were transfected with FLAG-SopF or control vector. We observed by western blot, that SopF expression was sufficient to inhibit ER-phagy under stimulated conditions (Fig. 4E, F). Consistently, RFP puncta formation was significantly decreased in cells transfected with HA-SopF compared to control cells (Supp Fig. 4B, C). Furthermore, SopF expression dramatically blocked endogenous FAM134B degradation by ER-phagy under stimulated conditions (Supp Fig. 4D). Together, these data indicate the bacterial effector SopF is required for Salmonella to inhibit ER-phagy.

A WT HEK293A cells expressing ss-RFP-GFP-KDEL were AA-starved for 6 h or 3 h, then infected with ST WT or ST ΔsopF for 3 h. ER-phagy was measured via ss-RFP-GFP-KDEL processing. Actin = loading control. NR nutrient rich; AA amino acids; ST = Salmonella Typhimurium. B Normalized Free RFP-Actin ratio from (A). Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001; ns no significance. C WT HEK293A cells expressing ss-RFP-GFP-KDEL were AA-starved for 6 h or 3 h + ST WT/ΔsopF infection for 3 h, fixed, and imaged. ss-RFP-GFP-KDEL = yellow; Free RFP = red. Scale bar = 5 μm. D Average RFP puncta/cell from (C). Data are presented as mean values ± SD of five biological experiments. ANOVA, ***P < 0.001. E WT cells from (A) transfected with FLAG-SopF or mock plasmid, AA-starved for 6 h or 3 h + ST WT infection for 3 h. ER-phagy measured. Actin = loading control. F Normalized Free RFP-Actin ratio from (E). Data are presented as mean values ± SD of three biological experiments. ANOVA, ***P < 0.001. G HEK293A FAM134B KO cells with ss-RFP-GFP-KDEL transfected with FAM134B WT, G216R, or FLAG-SopF, then AA-starved for 6 h. Vinculin = loading control. H Normalized Free RFP-Vinculin ratio from (G). Data are presented as mean values ± SD of five biological experiments. ANOVA, **P < 0.01; ***P < 0.001.
Because forcing FAM134B oligomerization with the G216R mutant could bypass Salmonella-mediated ER-phagy blockage, we sought to determine if SopF effects on ER-phagy could also be prevented by FAM134B G216R. Indeed, FAM134B KO cells co-expressing FLAG-SopF and FAM134B G216R were able to significantly induce ER-phagy when stimulated compared to cells transfected with FLAG-SopF and FAM134B WT (Fig. 4G, H). These data suggest Salmonella targets FAM134B oligomerization through SopF.
Recently, the crystal structure of SopF was solved revealing it to be a member of the ADP-ribosyltransferase superfamily28. Indeed, SopF has been shown to catalyze the transfer of ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to the v-ATPase subunit ATP6V0C leading to the inhibition of bacterial autophagy, but not general autophagy or other types of selective autophagy28,29. To test if SopF could directly interact with and target FAM134B, we co-immunoprecipitated FLAG-SopF and endogenous FAM134B, detecting a possibly transient interaction between both proteins (Fig. 5A). To further validate FAM134B and SopF interaction, we performed a TurboID assay, which relies on biotin-based proximity labeling and can detect transient interactions more reliably than co-IP. Briefly, SopF was tagged with a more promiscuous form of BirA, an Escherichia coli-derived biotin ligase. After incubation with biotin, proteins in the near vicinity of the TurboID-tagged SopF become biotinylated, enabling their identification after streptavidin pull-down30. We observed that FAM134B was dramatically enriched after streptavidin pull-down when TurboID-SopF was expressed compared to the TurboID control, indicating SopF comes in close proximity to FAM134B (Fig. 5B).
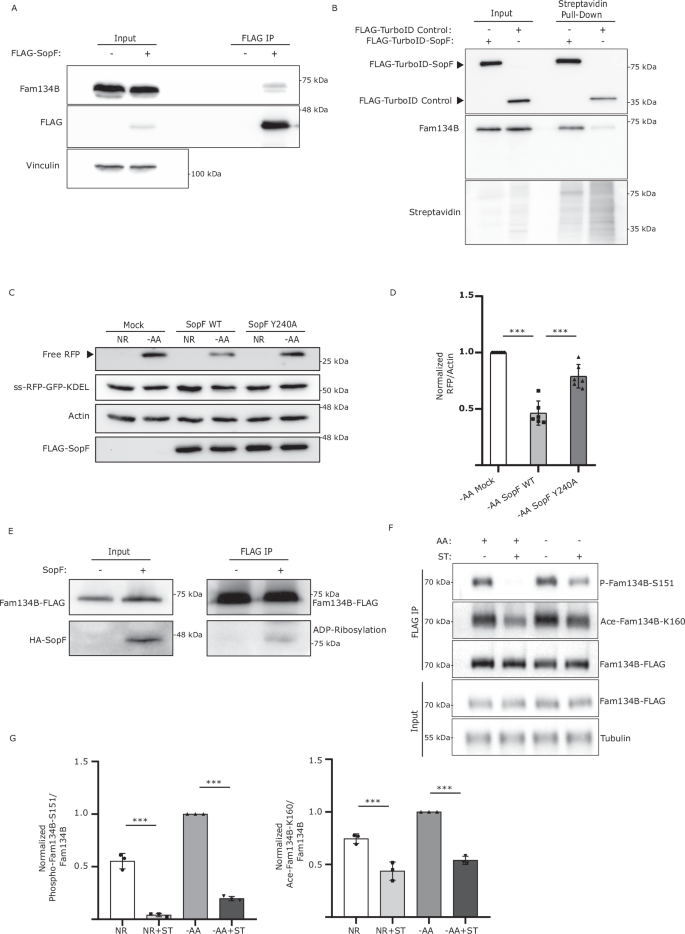
A WT HEK293A cells were transfected with FLAG-SopF or mock plasmid. FLAG-SopF was immunoprecipitated. Representative data from 3 biological experiment. B WT HEK293A cells transfected with FLAG-TurboID-SopF or control plasmid, incubated with Biotin, and subjected to streptavidin pull-down, representative images of 3 biological experiment. C WT HEK293A cells expressing ss-RFP-GFP-KDEL transfected with FLAG-SopF WT, Y240A, or mock plasmid, AA-starved for 6 h. ER-phagy was measured. Actin = loading control. D Normalized Free RFP-Actin ratio from (C). Data are presented as mean values ± SD of six experiments. ANOVA, ***P < 0.001. E FAM134B KO cells transfected with FAM134B-FLAG + HA-SopF or mock plasmid. FLAG was immunoprecipitated. ADP-ribosylation was detected using a pan-ADP ribose binding reagent. Representative image of five biological experiments. F HeLa cells were transfected with FAM134B-FLAG, AA-starved for 3 h and/or ST-infected for 3 h. FLAG was immunoprecipitated. Fam134B Ser151 phosphorylation and Lys160 acetylation were detected using specific antibodies. Tubulin = loading control. G Normalized Phospho-Fam134B-S151/Total Fam134B and Ace-Fam134B-K160/Total Fam134B ratios from (F) Data are presented as mean values ± SD of three biological experiments. ANOVA, ***P < 0.001. Source data are provided as a Source Data file.
SopF mutations preventing its binding to the N-ribose (E325A) or the nicotinamide (Y224A, Y240A) group of NAD+ largely blocked SopF ADP-ribosylation activity28. Consistently, SopF Y240A failed to prevent ER-phagy activation as observed by a significant increase in RFP production compared to SopF WT. (Fig. 5C, D). Additionally, SopF mutants E325A and Y224A also showed increased ER-phagy compared to SopF WT (Supp Fig. 4E and Supp Fig. 4F). Moreover, expression of SopF WT, but not the ADP-ribosylation mutant SopF Y240A, reduced the formation of FAM134B oligomers (Supp Fig. 4G). To test if SopF could ADP-ribosylate FAM134B we used a pan-ADP-ribose binding reagent capable of detecting both mono and poly-ADP ribosylation. A band was observed when FAM134B was IP in the presence of SopF, which was absent in the control IP, suggesting SopF may directly ADP-ribosylate FAM134B (Fig. 5E).
Recent studies have reported the importance of FAM134B acetylation at Lys160 and phosphorylation at residues Ser149, Ser151 and Ser153 in promoting its oligomerization and ER-phagy activation9,10. We hypothesized that one of the ways Salmonella could inhibit FAM134B oligomerization was by preventing these post-translational modifications. Mass spectrometry analysis of FAM134B in the absence of SopF, showed phosphorylation in Ser151, which was not detected when SopF was co-expressed (Supp Fig. 4H). Consistently, Salmonella infection significantly decreased both FAM134B Ser151 phosphorylation and Lys160 acetylation, in both nutrient rich and starvation conditions (Fig. 5F, G), indicating Salmonella directly targets FAM134B regulation and its ability to oligomerize. Further studies will be required to determine if Salmonella-mediated inhibition of FAM134B oligomerization is driven by SopF directly ADP-ribosylating FAM134B or upstream regulators. Together these results indicate that SopF ADP-ribosylation activity is required for ER-phagy inhibition.
FAM134B restricts Salmonella growth
To better understand the nature of FAM134B-mediated resistance to intracellular Salmonella, we next looked at the factors that could contribute to the FAM134B KO defect, such as bacteria vesicle escape, clearance, and growth. First, we infected WT and FAM134B KO cells with GFP Salmonella and quantified growth post-invasion for 4 hrs. To distinguish between external and internalized Salmonella, cells were stained with a LPS antibody before permeabilization, allowing selective labeling and exclusion of external bacteria in our quantifications. We observed a similar level of bacterial internalization in both WT and FAM134B KO at 1 h (Fig. 6A), indicating similar levels of infection rate. Interestingly, we observed a significant difference in growth rate between WT and FAM134B KO beginning at 2 h post infection that persisted through the remaining time points. Escape from Salmonella-containing vesicles (SCV) is known to increase the growth rate of intracellular Salmonella, which is significantly higher in the cytosol. However, SCV escape in WT cells typically occurs well after 4 h post infection31 and results in the growth of rod-shaped Salmonella that are more spread out than those growing in the SCV. Given the early timepoint of divergence in growth rates and the morphology of internalized Salmonella, it is highly unlikely that FAM134B KO defects are a result of early escape from the SCV. Nevertheless, we quantified the amount of cytosolic Salmonella compared to the total population using a chloroquine resistance CFU assay. Chloroquine accumulates in high concentration within endosomes, preferentially targeting vacuolar, rather than cytosolic Salmonella, rendering SCV-containing Salmonella transcriptionally inactive and non-replicative31. Consistent with other studies, we observed that cytosolic Salmonella accounted for 10% of total bacteria in infected WT cells at similar time points31 (Supp. Fig. 5A). However, there was no significant difference in cytosolic Salmonella between infected WT and FAM134B KO cells, indicating FAM134B is not involved in Salmonella escape from SCVs (Supp. Fig. 5A). We next sought to measure the impact of FAM134B on autophagic bacterial clearance, termed xenophagy. To estimate the relative contribution of FAM134B in Salmonella clearance compared to other forms of autophagy, we performed a CFU assay in WT and FAM134B KO cells treated with the VPS34 inhibitor, VPS34-IN1. The class-III phosphatidylinositol-3-phosphate kinase complex, which is formed by the catalytic subunit PIK3C3/VPS34, among others, plays an essential role in autophagy activation32, thus, VPS34 inhibition blocks general autophagy24. As expected, inhibiting VPS34 dramatically increased Salmonella levels in both WT and FAM134B KO cells. However, VPS34-IN1-treated FAM134B KO cells showed a significant increase in Salmonella burden compared to treated WT cells, suggesting FAM134B may have an additional role in preventing Salmonella growth, not directly related to xenophagy (Supp. Fig. 5B). Autophagosomal degradation of Salmonella is mediated by LC3 recruitment to bacteria and autophagy-deficient cells have been shown to be more permissive for Salmonella growth33. However, the impact of ER-phagy in Salmonella clearance is unclear. Strikingly, FAM134B KO cells contained less LC3B-positive Salmonella compared to WT cells, suggesting a role for FAM134B and ER-phagy in Salmonella clearance (Fig. 6B, C). LC3B targeting to Salmonella is modulated by various xenophagy adapters such as TBK1, which is involved in phosphorylating different xenophagy receptors and recruiting them to the surface of Salmonella for degradation34. Interestingly, we found no significant difference in TBK1 recruitment to Salmonella in WT and FAM134B KO cells (Supp Fig. 5C, D), suggesting FAM134B effects on Salmonella replication might be either downstream of TBK1, or not directly related to xenophagy.

A WT and FAM134B KO HEK293A cells were infected with ST for 1 h followed by Gentamicin wash-off. Cells were fixed at indicated time points. ST, Salmonella Typhimurium. Representative images are shown. ST, green; LPS, far red; DAPI; blue. Scale bar 10 μm. Data are presented as mean values ± SD of five biological experiments. The number of ST were calculated from a minimum of 100 cells. ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001. B HEK293A WT and FAM134B KO cells were infected for 1 h. Autophagic capture of ST was analyzed by immunostaining for LPS and LC3B. Representative images are shown. ST, green; LC3B, red; LPS, far red; DAPI; blue. Scale bar 5 μm. C Percentage of ST colocalizing with LC3B in WT and FAM134B KO cells from (B). Data are presented as mean values ± SD of five biological experiments. The percentage of colocalization was calculated from a minimum of 100 cells. Student’s t test, ***P < 0.001. D HEK293A WT and FAM134B KO cells were infected with either ST WT or ST ΔsopF for 1 h. Autophagic capture of ST was analyzed by immunostaining for LPS and LC3B. Representative images are shown. ST, green; LC3B, red; LPS, far red; DAPI; blue. Scale bar 5 μm. E The ratio between the number of ST colocalizing with LC3B and total internalized ST from (D) is presented. Data are presented as mean values ± SD of five biological experiments. LC3B-ST colocalization was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001; ns, no significance. F HEK293A WT and FAM134B KO were transfected with either FLAG-SopF or a mock plasmid followed by WT ST infection. Bacterial content was determined through colony forming unit (CFU). Data are presented as mean values ± SD of three biological experiments. ANOVA, **P < 0.01; ns, no significance. Source data are provided as a Source Data file.
To determine if SopF is required for the suppression of LC3-positive puncta in FAM134B KO cells, we quantified the localization of LC3B to SopF-deficient Salmonella. We observed the previously reported increase of LC3B colocalization to ΔsopF Salmonella compared to WT Salmonella in our control cells35. However, SopF deletion exerted no significant difference in LC3B colocalization in FAM134B KO cells (Fig. 6D, E). Similarly, SopF overexpression significantly increased WT Salmonella levels in control cells, while overexpression of SopF in FAM134B KO cells showed no significant difference in Salmonella viability compared to mock transfected FAM134B KO cells (Fig. 6F), further indicating that SopF effects on Salmonella growth are linked to FAM134B. Moreover, it reinforces our working model that SopF is inhibiting ER-phagy to influence ER morphology and promote intracellular Salmonella survival. This hypothesis was further corroborated by electron microscopy images showing increased ER area in cells stabily expressing SopF compared to control cells (Supp. Fig. 5E), indicating SopF-mediated inhibition of ER-phagy reconfigures ER morphology. The mechanisms that specifically link FAM134B-dependent ER-phagy to xenophagy are unclear at this time, however, these results suggest an additional, yet undiscovered, pathway linking FAM134B to Salmonella restriction.
Infected FAM134B KO mice are more susceptible to Salmonella infection
We next sought to determine the pathophysiological effects of Salmonella-mediated inhibition of ER-phagy in vivo. To this end, we performed oral gavage Salmonella infection of WT and FAM134B KO mice36 and analyzed bacterial burden and intestinal damage 5 days post infection. Histochemical analysis of hematoxylin and eosin (H&E) stained intestine samples of infected WT mice presented mucosa and submucosa infiltration, along with occasional damage (Fig. 7A). However, infected FAM134B KO mice showed severe wall, mucosa and submucosa infiltration, as well as marked necrotic damage, fibrin formation and edema. Following H&E, mucosa and submucosa samples were stained to detect Salmonella levels, which showed a significant increase in bacterial burden in FAM134B KO compared to WT cells (Fig. 7B, C). These results are consistent with our in vitro data, highlighting FAM134B role in restricting Salmonella growth. Infections were repeated as described above and WT and FAM134B KO Salmonella load was measured by CFU in feces, spleen, and intestine. Consistent with immunofluorescence analysis, we observed significantly higher levels of Salmonella in the tissue and feces of FAM134B KO mice (Fig. 7D, E, F). We next tried to determine if our previous results extended to primary macrophages, key agents involved in the innate and adaptive immune response against Salmonella infection. Thus, we measured FAM134B-mediated ER membrane scission in starved bone marrow derived macrophages (BMDM) obtained from WT mice that were infected with either Salmonella WT or ΔsopF. As expected, ER-phagy activation induced FAM134B puncta formation, which was significantly blocked by Salmonella WT, but not SopF defective Salmonella (Fig. 7G, H), recapitulating our results in endothelial cells. Finally, BMDM obtained from FAM134B KO mice displayed significantly more Salmonella than WT BMDM after infection (Fig. 7I), further underlining the importance of FAM134B in anti-bacterial response in multiple tissues and cell types. Overall, our results indicate that FAM134B is an important factor in controlling bacterial infection in vivo.

A WT and FAM134B KO C57BL/6J mice were infected with WT GFP ST through oral gavage and after 5 days, their small intestine was fixed and stained with H&E. (a) infiltration, (b) necrosis, (c) fibrosis, and (d) edema. Pathology scores of post infected tissues are presented. Data are presented as mean values ± SD of n = 4 mice. ANOVA, *P < 0.05; **P < 0.01. ST, Salmonella Typhimurium. Scale bar 50 μm. B WT and FAM134B KO mice small intestine samples from (A) were stained with DAPI and GFP to detect ST. ST, Red; DAPI, Blue. Scale bar: 5, 20 and 50 μm. C The number of cells infected with ST from (B) were quantified. Data are presented as mean values ± SD of n = 4 mice, ST infection was calculated from a minimum of 350 cells. Student’s t test, ***P < 0.001. WT and FAM134B KO mice were infected with ST through oral gavage and after 5 days their spleen, whole intestine and feces were collected. Bacterial content was determined through colony- forming unit (CFU). Data are presented as mean values ± SD (D, E) n = 7 mice and (F) n = 7 biological experiment. Student’s t test, *P < 0.05; **P < 0.01. G BMDM, bone marrow derived macrophages from WT mice were kept in NR media, AA starved for 2 hr or starve for 1 h followed by ST WT or ST ΔsopF infection in starvation media for an additional 1 h. Starved samples were treated with BafA1. BMDM were then fixed and imaged by confocal microscopy. Representative images are shown. NR Nutrient Rich, AA amino acids, BafA1 Bafilomycin A1. Scale bar 5 μm. H Average number of Fam134B puncta per cell from (G) were quantified. Data are presented as mean values ± SD of five biological experiments. Average Fam134B puncta formation was calculated from a minimum of 100 cells. ANOVA, ***P < 0.001. I BMDM from WT and FAM134B KO mice were infected with ΔinvA ST. Bacterial content was determined through colony-forming unit (CFU). Data are presented as mean values ± SD of three biological experiments. Student’s t test, *P < 0.05. Source data are provided as a Source Data file.




